Assessing Duration Discrimination: Psychophysical Methods and Psychometric Function Analysis
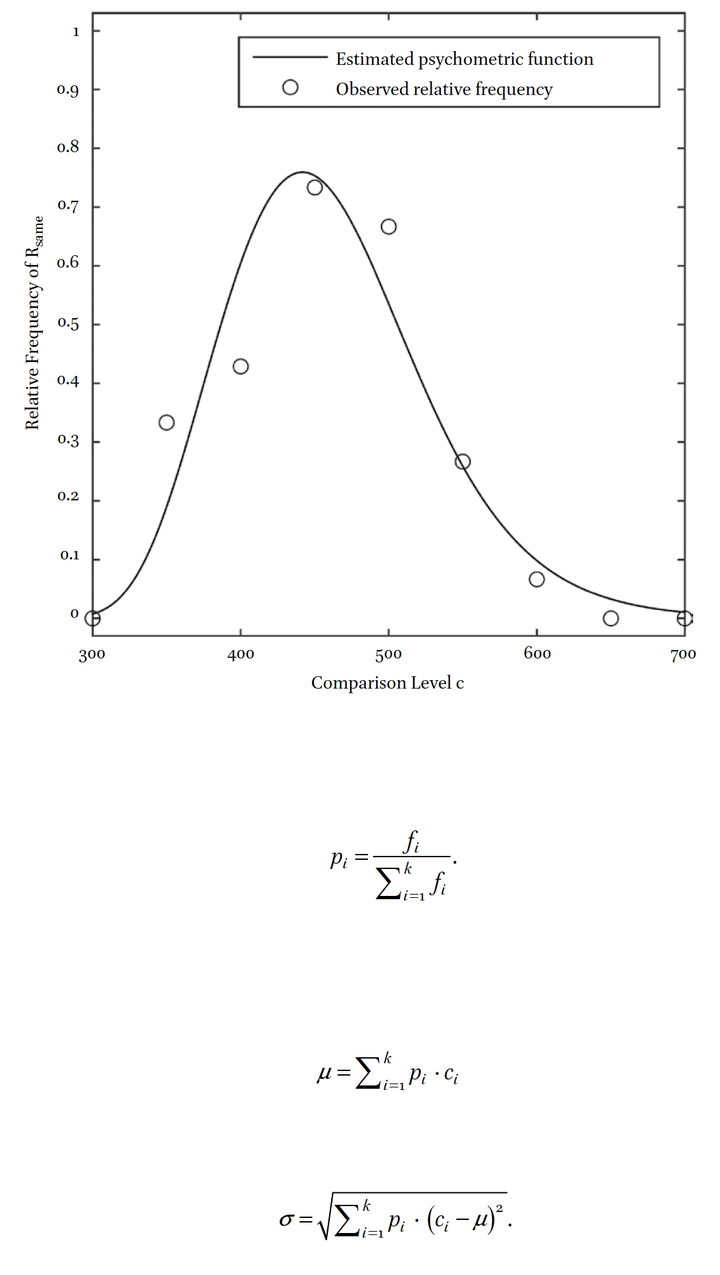
Abstract
Predicting the binding mode of flexible polypeptides to proteins is an important task that falls outside the domain of applicability of most small molecule and protein−protein docking tools. Here, we test the small molecule flexible ligand docking program Glide on a set of 19 non-$α$-helical peptides and systematically improve pose prediction accuracy by enhancing Glide sampling for flexible polypeptides. In addition, scoring of the poses was improved by post-processing with physics-based implicit solvent MM- GBSA calculations. Using the best RMSD among the top 10 scoring poses as a metric, the success rate (RMSD ≤ 2.0 Å for the interface backbone atoms) increased from 21% with default Glide SP settings to 58% with the enhanced peptide sampling and scoring protocol in the case of redocking to the native protein structure. This approaches the accuracy of the recently developed Rosetta FlexPepDock method (63% success for these 19 peptides) while being over 100 times faster. Cross-docking was performed for a subset of cases where an unbound receptor structure was available, and in that case, 40% of peptides were docked successfully. We analyze the results and find that the optimized polypeptide protocol is most accurate for extended peptides of limited size and number of formal charges, defining a domain of applicability for this approach.